In flies, loss of function mutations in genes encoding components of Polycomb Repressive Complex 2 (PRC2)—an H3K27me3 specific methyltransferase complex—result in decreased levels of H3K27me3 and increased lifespan
PTM crosstalk relevant to longevity is histone bivalency where two histone PTMs that normally oppose each other (H3K4me3 and H3K27me3) act synergistically in combination to promote a unique intermediate state of chromatin that appears “poised” for gene activation
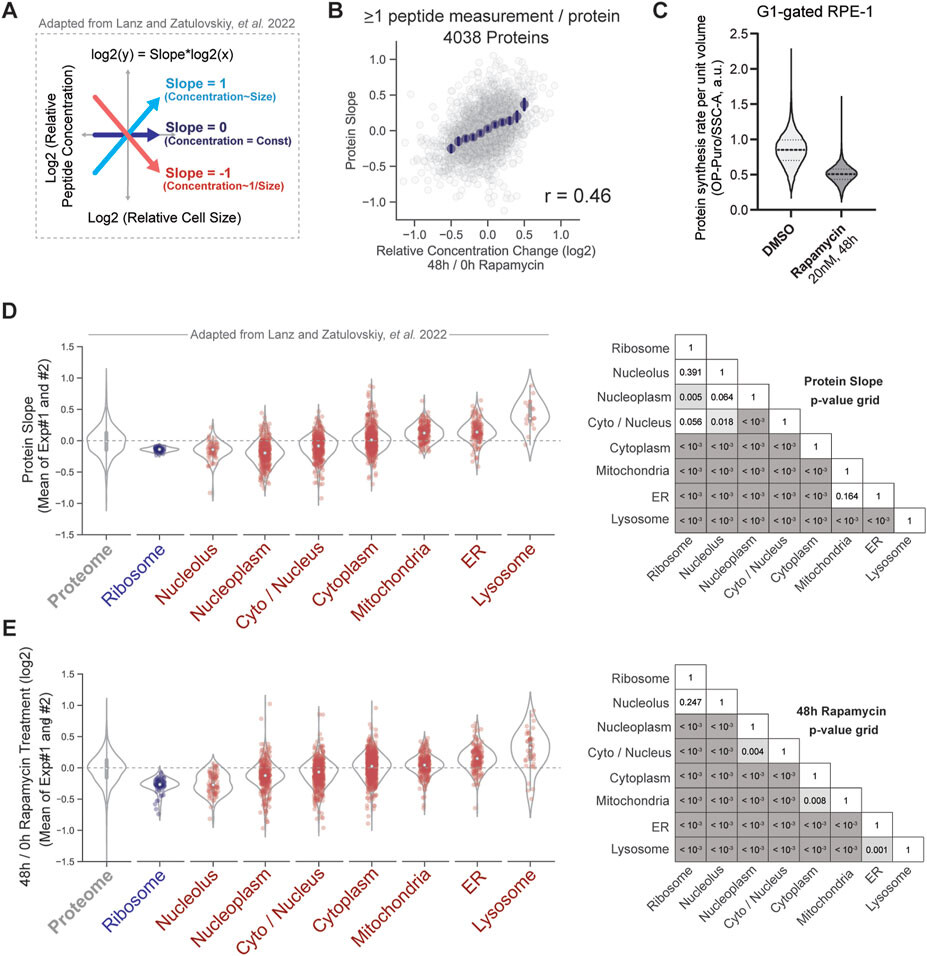
SIRT6 T294 phosphorylation site is particularly interesting given the role of SIRT6 in regulating lifespan. A more thorough phylogenetic analysis of T294 reveals that it is only present in a few long-lived groups such as old-world primates, cetaceans, beavers, tree squirrels, as well as David’s myotis bat; however, the majority of mammals appear to lack this site. Consistent with its potentially important role, phosphorylation of T294 is the most highly observed modification of human SIRT6 (cataloged in the PhosphoSitePlus database);[94] yet, several studies failed to reveal its role. T294 resides outside of the highly conserved sir-tuin catalytic domain within a proline-rich region that bridges to a less conserved C-terminus that may facilitate protein–protein interactions.[95] Substitution of T294 with glutamic acid (T294E) that in many cases would mimic a constitutive phosphorylation did not affect SIRT6 nuclear (or nucleolar) localization.[96] Similarly, alanine substitution (T294A) did not reduce SIRT6’s ability to stimulate DNA repair after oxidative stress
Since PTMs are not directly genetically encoded, engineering these features in vivo was beyond reach until recently. Novel engineering strategies such as protein ligation/splicing, non-natural amino acid mutagenesis, or inactive CRISPR/Cas9-fusion protein-based localization of epigenetic modifiers may permit the engineering of designer PTMs/proteoforms in vivo.[104] We expect that future proteomics studies of longevity will incorporate these approaches to correlate PTMs/proteoforms with lifespan.
Moreover, an AP site is a location in DNA that has neither a purine or a pyrimidine base due to DNA damage, they are the most prevalent type of endogenous DNA damage in cells. AP sites can be generated spontaneously or after the cleavage of modified bases, like 8-OH-Gua
Moreover, oxidative DNA damage like 8-oxo-dG contributes to carcinogenesis through the modulation of gene expression, or the induction of mutations.[54] On the condition that 8-oxo-dG is repaired by BER, parts of the repair protein is left behind which can lead to epigenetic alterations, or the modulation of gene expression
The term G4 DNA was originally reserved for these tetramolecular structures that might play a role in meiosis.[5] However, as currently used in molecular biology, the term G4 can mean G-quadruplexes of any molecularity. Longer sequences, which contain two contiguous runs of three or more guanine bases, where the guanine regions are separated by one or more bases, only require two such sequences to provide enough guanine bases to form a quadruplex. These structures, formed from two separate G-rich strands, are termed bimolecular quadruplexes. Finally, sequences which contain four distinct runs of guanine bases can form stable quadruplex structures by themselves, and a quadruplex formed entirely from a single strand is called an intramolecular quadruplex.[17]
#bionumbers
Guanine (G) bases in G-quadruplex have the lowest redox potential causing it to be more susceptible to the formation of 8-oxoguanine (8-oxoG), an endogenous oxidized DNA base damage in the genome. Due to Guanine having a lower electron reduction potential than the other nucleotides bases,[53]8-oxo-2’-deoxyguanosine (8-oxo-dG), is a known major product of DNA oxidation
#DNArepair
AP site damage was found to be predominant in PQS regions of the genome, where formation of G-quadruplex structures is regulated and promoted by the DNA repair process, base excision repair (BER).[49] Base excision repair processes in cells have been proved to be reduced with aging as its components in the mitochondria begin to decline, which can lead to the formation of many diseases such as Alzheimer’s disease (AD).[50] These G-quadruplex structures are said to be formed in the promoter regions of DNA through superhelicity, which favors the unwinding of the double helical structure of DNA and in turn loops the strands to form G-quadruplex structures in guanine rich regions.[51] The BER pathway is signalled when it indicates an oxidative DNA base damage, where structures like, 8-Oxoguanine-DNA glycosylase 1 (OGG1), APE1 and G-quadruplex play a huge role in its repair. These enzymes participate in BER to repair certain DNA lesions such as 7,8-dihydro-8-oxoguanine (8-oxoG), which forms under oxidative stress to guanine bases.[52]