featured in recent ARDD2023 video, this goes down with aging (Npc1 in yeast)
https://www.nature.com/articles/s41586-023-06802-1
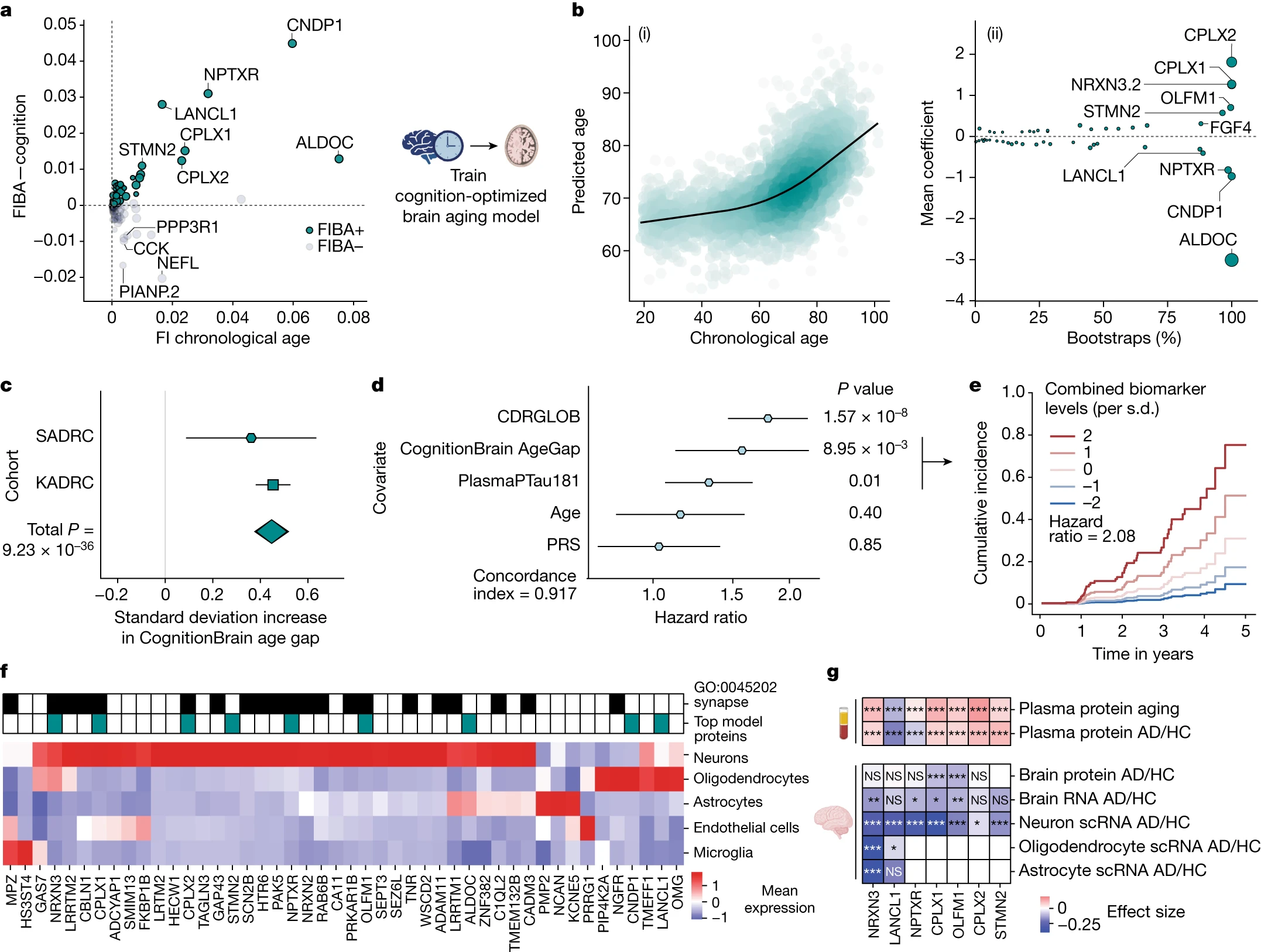
Given the significant associations between the CognitionBrain age model and several brain aging metrics, we sought to uncover new insights into brain aging mechanisms by examining the proteins that make up the model. A total of 47 of the 49 model proteins were detectable in human brain single-cell RNA sequencing (scRNA-seq) data and most could be mapped to neurons and glia with high specificity (Fig. 3f). Proteins with the largest positive weights in the model (Fig. 3c) included the synaptic proteins complexin 1 (CPLX1), complexin 2 (CPLX2) and neurexin 3 (NRXN3)—which all have genetic links to cognition and AD31,32,33—and stathmin 2 (STMN2) and olfactomedin 1 (OLFM1)—which are involved in neurite outgrowth and axon growth cone collapse34,35. Proteins with large negative weights in the model such as Aldolase Fructose-Bisphosphate C (ALDOC), neuronal pentraxin receptor (NPTXR), carnosine dipeptidase 1 (CNDP1) and Lanc Like Glutathione S-Transferase 1 (LANCL1). ALDOC, NPTXR and CNDP1 are expressed in astrocytes, neurons and oligodendrocytes, respectively (Fig. 3f) and have been proposed as CSF biomarkers for AD36,37. LANCL1, which is primarily expressed in oligodendrocytes (Fig. 3f), has been shown to be crucial for neuronal health in mouse models38. The model also implicated alterations in the glycosylated extracellular matrix through the proteins tenascin R (TNR), neurocan (NCAN) and heparan sulfate-glucosamine 3-sulfotransferase 4 (HS3ST4), underlining the role of the extracellular matrix in brain aging.
Paraoxonase 1 (PON1) is one of most studied genes associated with cardiovascular disease, oxidative stress, inflammation, and healthy aging. Specifically, PON1 plays an important role in detoxifying organophosphorus compounds and removing harmful oxidized lipids 44. The genetic variant of PON1 (R192Q) significantly decreases PON1 activity and is known to be associated with an increased risk of cardiovascular disease and neurodegenerative diseases 45. Interestingly, the PON1 Q allele is significantly depleted in centenarians 45. We analyzed the relationship between PON1 activity and epigenetic age in 48 whole blood samples (Fig. 6a) 46. DamAge shows a significant negative correlation with PON1 activity (R = −0.55, p = 0.0062), whereas AdaptAge showed a significant positive correlation with PON1 activity (R = 0.69, p = 0.0003). Again, this association was not observed by other epigenetic clocks, except for Horvath age, but with a less significant negative correlation (P = 0.04). Together, we showed that DamAge can reliably detect damage-related biological age acceleration.
2.4.1 Dissipative and Conservative Genes
Specifically, our analysis revealed distinct examples of these categories. Genes such as MKRN1,
SESN1, and ATR were identified as conservative, as their embeddings remained stationary in the
embedding space across all stages of life. Conversely, genes such as ALDH3B1, NR2C2, and HERPUD1 displayed higher drift from their initial embeddings, categorizing them as dissipative (Figure
5a). Interestingly, the classification of genes as conservative or dissipative appears independent of
their biological roles. Both types of genes are involved in a wide range of cellular processes, including
DNA repair, stress response, and oxidative metabolism. This observation suggests that conservation
and dissipation are universal characteristics intrinsic to the aging process and occur across diverse
molecular pathways. This approach allows us to move beyond static measures of gene expression and
explore the broader molecular interplay that defines biological aging.
Our analysis revealed alterations in the drift of gene embeddings under diseased conditions, a fundamental shift in gene regulatory dynamics. Specifically, the ATR gene, typically characterized as a
conservative gene with stable embeddings in healthy cells, exhibited a marked increase in drift within
diseased samples (Figure 5b). This shift suggests that ATR, which plays a critical role in DNA damage response and cell cycle regulation, may adopt a different expression pattern under pathological
conditions. A similar pattern was observed for the SAFB2 gene, a regulator involved in chromatin
organization and stress response, which also displayed increased drift in disease states. These findings
could imply a broader trend where genes with stable behavior in healthy tissues become destabilized
in the presence of disease. Interestingly, the TDP2 gene, classified as a dissipative gene due to its
high variability in healthy samples, demonstrated an opposite behavior, maintaining a conservative
embedding in diseased conditions (Figure 5b). This unexpected stability could suggest a pot
===
from some of the latest paper
Here’s a compact, mechanism-focused rundown of each item you listed—and why perturbing it can globally rewire modules across tissues.
PCDHG (protocadherin-γ cluster)
What they are: A tandem gene cluster (~22 variable exons spliced to 3 constant exons) on 5q31; part of the clustered protocadherins (PCDHA/PCDHB/PCDHG). Each neuron stochastically expresses a subset, generating a quasi-unique surface “barcode.”
Protein/function: Type-I cadherin-superfamily cell-adhesion receptors with extracellular cadherin repeats mediating strict homophilic binding; intracellular tails connect to cytoskeleton and signaling (e.g., PKC, FAK).
Biological roles:
- Neuronal self-avoidance and tiling, dendritic arborization, synapse stabilization—key for circuit assembly.
- Contribute to neuronal identity and neurite sorting; loss yields exuberant self-crossing and synaptic defects.
- Expressed outside CNS at lower levels; roles in epithelial adhesion and cancer cell contacts reported.
Regulation: A CTCF/cohesin-organized locus with promoter choice controlled by chromatin looping and enhancer hubs; activity-dependent and developmental cues reshape expression.
Why a “global disruptor”: Changing PCDHG dosage/patterns alters cell–cell recognition logic and tissue architecture; because the locus is chromatin-architected, perturbations ripple through 3D genome contacts, shifting co-expression/interaction modules broadly in neurogenic and epithelial tissues.
MEST (PEG1; Mesoderm-Specific Transcript)
What it is: An imprinted gene on 7q32.2—paternally expressed, maternally methylated. Encodes a secreted or ER-localized α/β-hydrolase–fold protein (Ser-His-Asp catalytic triad).
Biological roles:
- Placental and embryonic growth, mesenchymal lineage programs.
- Adipogenesis & metabolism: modulates Notch and Wnt signaling; MEST upregulation promotes adipocyte hypertrophy and insulin-resistance phenotypes in models.
- Vascular/mesenchymal remodeling; frequently dysregulated in cancers (epithelial–mesenchymal transition, invasion).
Regulation/clinical: Sensitive to imprinting/epigenetic state; aberrant methylation linked to growth-restriction syndromes and tumorigenesis.
Why a “global disruptor”: Because imprinting sets system-wide dosage, MEST shifts mesenchymal–epithelial balance, metabolic signaling (Notch/Wnt), and secreted cues across tissues—repositioning modules that couple development, metabolism, and stromal remodeling.
HDAC4 (Histone Deacetylase 4; class IIa)
- What it is: A class IIa HDAC that shuttles between cytoplasm and nucleus; CaMK-dependent phosphorylation recruits 14-3-3 to export; catalytic pocket is weak, but HDAC4 acts as a scaffold recruiting HDAC3/NCoR/SMRT.
- Transcriptional targets: Potent repressor of MEF2-dependent programs (myogenesis, synaptic plasticity), and modulates RUNX2/SOX9 in chondrogenesis; affects neuronal survival, learning, and stress responses.
- Development/disease: Dosage changes cause skeletal patterning defects (e.g., brachydactyly mental retardation in 2q37 del), cardiomyocyte hypertrophy, neurodevelopmental phenotypes; HDAC4 is a major epigenetic node responsive to calcium signaling.
- Why a “global disruptor”: As a signal-responsive chromatin hub, HDAC4 reprograms acetylation landscapes and MEF2/RUNX networks across multiple tissues, shifting module boundaries that link signaling → chromatin → transcription.
HOX family (HOXA/B/C/D clusters)
What they are: 39 homeobox transcription factors arranged in four clusters (HOXA–D) with spatiotemporal colinearity; master regulators of anterior–posterior patterning.
Biological roles:
- Embryonic patterning: axial identity, limb bud and organ field specification; RA (retinoic acid) and other morphogens gate their activation.
- Adult functions: maintain stem/progenitor cell states and positional identity in hematopoietic, epithelial, and mesenchymal niches.
- Cancer: context-dependent oncogenes/tumor suppressors; reactivation rewires cell identity programs.
Regulatory architecture: Clustered within TADs with long-range enhancers; dose and combination (“HOX code”) determine fate in many tissues.
Why a “global disruptor”: HOX factors sit atop cell-identity hierarchies; perturbing them resets gene-regulatory modules (adhesion, ECM, morphogenesis, metabolism) across diverse tissues, not just one lineage.
One-liner synthesis
- PCDHG perturbs cell-surface code → tissue wiring/architecture.
- MEST perturbs imprinted dosage → mesenchymal/metabolic signaling.
- HDAC4 perturbs signal-to-chromatin coupling → broad transcriptional states.
- HOX perturbs positional identity master switches → multi-tissue fate programs.
That combination—adhesion identity (PCDHG), imprinted metabolic/stromal control (MEST), epigenetic gating (HDAC4), and master positional TFs (HOX)—is exactly the sort of set that can reshuffle module arrangement across many tissues when you nudge any one member. If you want, I can map these to the specific module shifts reported in your Supplementary Table 5 (e.g., which co-expression blocks moved in which tissues).
Key disruptors, including PCDHGA1, MEST, HDAC4, and HOX genes, exacerbated aging signals across tissues. Notably, a resilient module enriched for NAD⁺ salvage metabolism supports therapeutic targeting of NAD⁺ in aging. PCDHGA1 emerged as a conserved cross-tissue driver, suggesting protocadherin-mediated adhesion plays a broader role in maintaining structural and signaling stability in multiple organ systems
DNA Methylation Ageing Atlas Across 17 Human Tissues | Research Square
Genes That Repair Early Glucose-Derived Damage
Early glucose-induced damage involves the formation of Schiff bases, Amadori products (fructosamines), and finally irreversible Advanced Glycation End-products (AGEs).
- Fructosamine-3-Kinase (FN3K): Catalyzes phosphorylation of Amadori (fructosamine) products, which starts their removal, reversing the early stages of glycation.
- DJ-1 (PARK7): Acts as a deglycase, removing methylglyoxal- and glyoxal-glycated adducts found in cysteine, arginine, and lysine.
- Transglycation system: Employs molecules such as glutathione and carnosine to cleave glycation intermediates, restoring protein function in a non-enzymatic process, especially for early Amadori products.
- Glyoxalase system (GLO1 and GLO2): Detoxifies reactive aldehydes (glyoxal, methylglyoxal) before they attach to proteins, preventing early glycation.
Summary Table
Enzyme/Gene Function Damage Step Repaired GPX1, GPX3 Peroxide reduction Prevents oxidative stress SOD1, SOD2 Superoxide detoxification Prevents oxidative stress TXN/TXNRD1 Repairs oxidized cysteines Disulfide reduction PRDX1, PRDX3 Repairs peroxidized proteins Peroxide detoxification CAT H2O2 elimination Prevents protein oxidation OXR1 Mitigates general oxidative damage Prevents neurodegeneration DJ-1/PARK7 Deglycase: Repairs early glycation Amadori/fructosamine adducts FN3K Removes fructosamine adducts Early glycation (Amadori) Transglycation Non-enzymatic reversible glycation repair Amadori products GLO1, GLO2 Detoxify glycation precursors Glyoxal, methylglyoxal Repair capacity for the first steps of glucose-derived protein damage centers on DJ-1, fructosamine-3-kinase, and the glyoxalase system, alongside broad antioxidant defense genes like GPX, SOD, and thioredoxin. These genes and enzymes collectively minimize, repair, or reverse early protein damage from both oxidative stress and glycation.
PSMB7
The genes encoding proteasome subunits—such as the beta7 (PSMB7) repeat—are crucial for proteasome core particle assembly and function in protein degradation and quality control. The human PSMB7 gene (proteasome 20S subunit beta type-7) is located on chromosome 9q34.11–q34.12 and encodes one of the seven beta subunits in the 20S core of the proteasome.
Key Proteasome Genes
- PSMB7: Encodes the beta7 subunit, which forms part of the proteolytic chamber in the 20S core particle. It has “trypsin-like” activity, cleaving after basic residues.
- Other beta subunit genes: PSMB1–PSMB6, along with PSMB7, collectively form the two inner heptameric rings of the proteasome’s 20S core.
- Alpha subunit genes: PSMA1–PSMA7 encode the outer rings of the 20S proteasome.
- Inducible subunits (immunoproteasome): Subunits such as PSMB8 (beta5i), PSMB9 (beta1i), and PSMB10 (beta2i) replace their constitutive counterparts in response to immune signals.
All cellular proteins tagged for degradation by ubiquitin are processed by the 26S proteasome, which contains the 20S core formed by these alpha and beta subunits, including beta7. The integrity and activity of these genes, especially PSMB7, are critical for maintaining protein homeostasis and cell viability.
SRSF1 (Serine/Arginine-Rich Splicing Factor 1) is an important splicing factor involved in mRNA processing, cellular senescence, and age-related phenotypes in humans, but neither cCREs (candidate cis-Regulatory Elements) nor CpG methylation patterns within SRSF1 are considered central determinants of human longevity for several reasons.pmc.ncbi.nlm.nih+2
Why SRSF1 cCREs/CpGs Aren’t Top Longevity Hits
- Redundancy and Complexity: SRSF1 is one member of a large family of splicing regulators whose functions overlap and are compensated by other splicing factors in aging and tissue homeostasis. Thus, even if SRSF1 is important, it’s not uniquely essential among hundreds of aging-related loci.frontiersin+1
- Indirect Effects: While SRSF1 influences splicing of various age-sensitive mRNAs (e.g., Bcl2L12, endoglin/ENG, LMNA), its impact is through gene networks rather than directly controlling lifespan or disease risk via its own regulatory cCREs or CpG methylation.pmc.ncbi.nlm.nih+1
- Association, Not Causation: Although SRSF1 expression declines with age and its overexpression can rejuvenate transcriptomic profiles or improve markers of senescence in culture, most large-scale human longevity GWAS do not identify SRSF1 regulatory variants (in cCREs or CpGs) as top longevity loci—likely because its contribution, although important, is not as impactful or directly targetable as genes involved in DNA repair, immune regulation, or metabolism.biorxiv
- Epigenetic Regulation: CpG methylation of SRSF1 might contribute to individual variation in aging or disease risk, but no strong evidence shows SRSF1 methylation as a major driver of longevity in large human cohorts.pmc.ncbi.nlm.nih+1
Summary Points
- SRSF1 is important for cellular youthfulness, splicing control, and stress resistance, but not the singular or dominant gene in longevity pathways.biorxiv+1
- cCREs and CpG sites in SRSF1 are not known to strongly control human lifespan in population studies, possibly due to redundancy or the polygenic/tissue-specific nature of splicing factor networks in aging.pmc.ncbi.nlm.nih+1
Other genes with direct impacts on genome stability, immune regulation, or metabolic adaptation are more consistently implicated as major “longevity genes” in humans.biorxiv
- The Splicing Factor SRSF1 as a Marker for Endothelial Senescence - PMC
- Frontiers | The Splicing Factor SRSF1 as a Marker for Endothelial Senescence
- Transcriptomic reprogramming screen identifies SRSF1 as rejuvenation factor | bioRxiv
- Inside the genome: understanding genetic influences on oxidative stress - PMC
- Splicing factor SRSF1 attenuates cardiomyocytes apoptosis via regulating alternative splicing of Bcl2L12 - PMC
- Changes in splicing factor expression are associated with advancing age in man - PMC
- https://www.sciencedirect.com/science/article/abs/pii/S0161589022004606
- https://www.sciencedirect.com/science/article/abs/pii/S152166162200122X
- https://aacrjournals.org/cancerdiscovery/article/13/7/1678/727620/Splicing-Factor-SRSF1-Promotes-Pancreatitis-and
- SRSF1 inhibits autophagy through regulating Bcl-x splicing and interacting with PIK3C3 in lung cancer - PMC
- Knockdown of the fly spliceosome component Rbp1(orthologue of SRSF1) extends lifespan | bioRxiv
Protein repair genes—such as those encoding methionine sulfoxide reductases, protein-L-isoaspartate methyltransferase, and deglycase enzymes—are not among the genes most strongly associated with exceptional human longevity in large-scale genetic studies. This is mainly because longevity in humans is influenced by a highly polygenic, redundant, and environmentally modulated network of mechanisms, rather than a reliance on a small set of uniquely important “repair” genes.
Reasons for Weak Association
- Redundancy: Many cellular pathways (e.g., chaperones, proteasomes, autophagy) overlap in maintaining protein quality, so no single repair gene typically controls lifespan on its own.
- Polygenic Traits: Longevity is regulated by hundreds of small-effect genetic variants across the genome, with larger effects often seen in genes regulating metabolism, stress response, and genome stability instead of just protein repair.
- Environmental and Lifestyle Influence: Diet, exercise, and exposure to toxins or infections heavily affect protein damage and repair; thus, genetic effects can be masked or diluted by these external factors in human populations.
- Survival Thresholds: Basic protein repair function is essential for cell viability, but once sufficient capacity is present, further increases may have diminishing returns for lifespan, especially in the presence of systemic failures linked to other pathways (e.g., immune aging, cardiovascular health).
What Genes Are More Commonly Linked with Longevity?
Genes more consistently associated with extended human lifespan include those tied to insulin/IGF-1 signaling (e.g., FOXO3), cholesterol transport (APOE), DNA damage repair (TP53, WRN), and inflammation—rather than those involved exclusively in direct protein repair.
Overall, while protein repair genes are key for cell survival and healthy aging, achieving exceptional human longevity depends much more on overall genomic stability, metabolic regulation, and robust systemic responses than on upregulation of any single protein repair gene.
Cell Junction/Membrane Stability in NMRs
- Tight Junction Proteins: NMRs display higher levels and unique distributions of major tight junction proteins (e.g., claudins, occludin, ZO-1) in gut and skin, reducing tissue permeability and maintaining robust barriers throughout lifespan.
- Adherens Junctions & Desmosomes: Their tissues show consistently elevated expression of proteins like E-cadherin, desmoglein, and desmocollin, supporting strong intercellular adhesion and resistance to detachment or inflammation.
- Membrane Lipid Composition: NMR cell membranes, especially in the brain, are exceptionally rich in cholesterol and possess short, saturated sphingomyelin chains. This results in stiffer, more ordered membranes that are less susceptible to oxidative damage and leakage, offering greater protection against age-related membrane dysfunction.
- Goblet Cells and Mucus: NMR intestines have more goblet cells and much higher mucus content, providing an even thicker, harder-to-penetrate physical barrier against irritants and pathogens.
- Longevity-Associated Barrier Genes: Transcriptome studies show increased expression of longevity and tumor suppressor genes, such as Cdkn1a, Cdkn2c, Igfbp3, and Ing2, in aged NMR skin and epithelial tissues, which bolster cellular boundary function and stress resistance.
Do NMRs Have Tighter Boundaries?
Yes—empirical measurements show NMRs have lower intestinal permeability (higher transepithelial electrical resistance), thicker gut and skin barriers, more stable cell junction complexes, and membranes more resistant to environmental and internal damage than mice and other rodents. This is considered a key adaptation for their extraordinary health span.
Feature NMR Adaptation Reference Tight junctions Claudin, occludin, ZO-1 upregulation Adherens/desmosomes Elevated E-cadherin, desmoglein Cholesterol-rich membranes Stiffer, less oxidizable lipid bilayers Goblet cells/mucus More cells, higher mucus barrier Barrier genes Cdkn1a, Cdkn2c, Igfbp3, Ing2 up These adaptations, together with DNA repair and proteostasis, are key factors in NMR longevity and resistance to common age-related pathologies.
One example is fructosamine-3-kinase (FN3K), an intracellular deglycation enzyme, and another is PIMT (protein L-isoaspartate O-methyltransferase), which repairs isoaspartyl damage in proteins. Both act primarily inside cells. Even when PIMT is released during injury, most extracellular damage remains out of reach, and a large share of this kind of damage ends up accumulating in ECM proteins.
PPT1 (Palmitoyl-protein thioesterase 1) is a lysosomal enzyme responsible for breaking down lipid-modified proteins